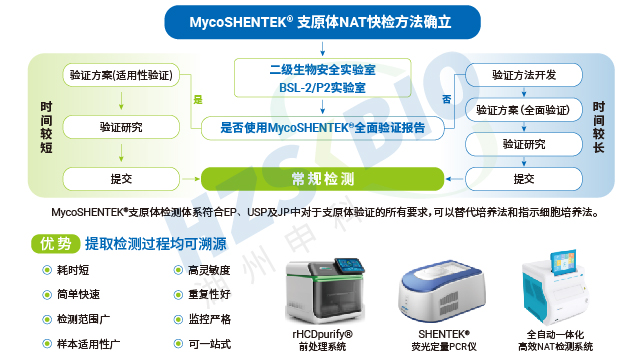
外源因子全自动核酸检测分析系统系统在数据追溯与合规管理方面进行了针对性设计,完美适配生物药行业的严格监管要求。系统内置三级权限管理机制,可对操作人员、检测项目、数据访问进行准确管控,同时具备完善的日志审计追踪功能,详细记录检测全流程的关键信息,确保数据可追溯、可核查,完全符合 21CFR Part11 法规要求。数据传输方面,系统支持与 LIMS 实验室信息管理系统无缝对接,实现检测数据的自动同步与集中管理,同时配备 USB 接口,可直接连接打印机打印实验结果,满足企业纸质记录存档需求。这些功能让支原体检测的每一个环节都处于合规管控之下,为企业应对监管检查、保障数据真实性提供了有力支持。
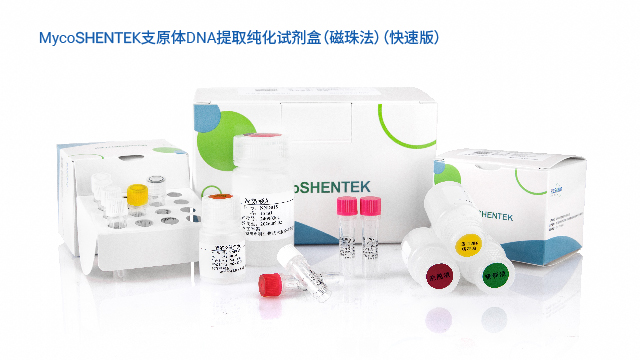
湖州申科支原体检测试剂盒通过FDA DMF备案,支持全球药品申报使用。河南复杂基质支原体检测试剂盒
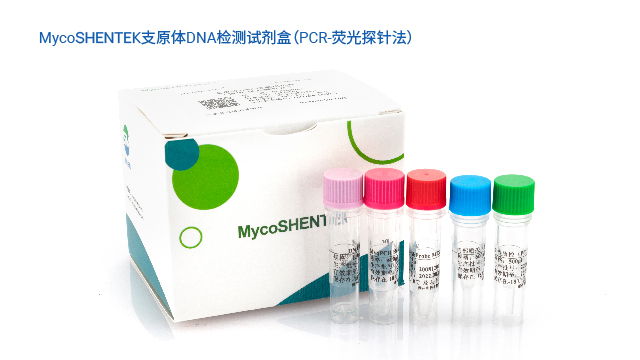
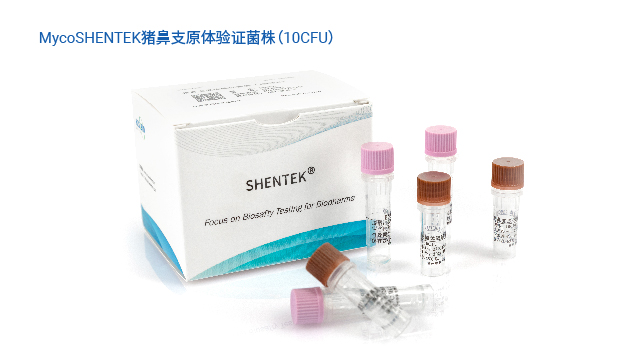
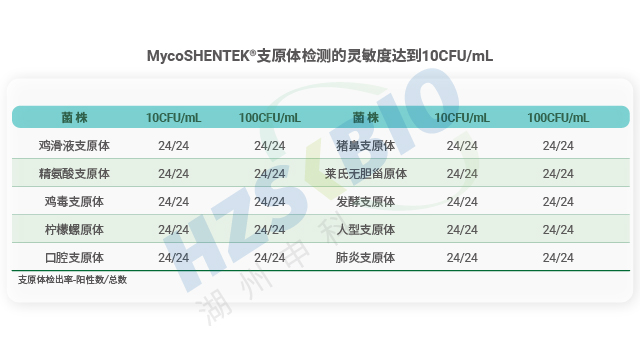
为解决传统方法的局限,各国药典纷纷认可支原体核酸检测(NAT)法作为替代方案。EP 2.6.7、USP <77>、JP <G3-14-170>均明确了 NAT 法的应用标准,《中国药典》3301 也规定 “可采用经国家药品检定机构认可的其他方法”,并明确 NAT 法替代培养法时检测限需达 10 CFU/mL,替代指示细胞培养法时需达 100 CFU/mL。NAT 法凭借检测速度快、特异性强的优势,成为新型生物制品支原体检测的理想选择,且法规明确只要通过相关验证,即可正式替代传统方法,为企业提供了合规且高效的检测路径。
河南细胞疗法产品支原体检测核酸扩增法支原体检测过程中,每批次培养基需做灵敏度测试,确保检测有效。
AdvSHENTEK外源因子全自动核酸检测分析系统凭借优越性能,为支原体检测提供坚实技术支撑。硬件方面,系统温度运行精度≤0.5℃,温度波动控制在 ±0.5℃以内,荧光强度 CV≤3%,确保检测结果的重复性与准确性;4 通道单独运行且支持同步检测,通道可叠加延展,兼顾检测效率与灵活性。软件方面,系统具备三级权限管理、日志审计追踪功能,完全符合 21CFR Part11 法规要求,支持 LIMS 系统连接、USB 数据导出及打印机直接打印,满足企业合规追溯需求。此外,系统运输与储存便捷,仪器可常规运输,检测试剂盒在 2-8℃环境下即可稳定保存,无需特殊冷链条件,进一步提升了产品的实用性与适配性。
湖州申科的支原体检测方案已在多个领域积累了丰富的客户申报案例,覆盖细胞疗法、抗体药物、疫苗、CRO/CDMO 等细分赛道。在细胞疗法领域,方案成功应用于已上市药物的方法变更,替换进口试剂盒并通过中检院复核,验证结果获得 CDE 认可用于产品放行检测在抗体药物领域,支持多家企业完成 BLA 申报;在疫苗与 CRO/CDMO 领域,为诸多头部企业提供 IND 申报支持。方案的应用场景涵盖从 IND 到 BLA/NDA 的全申报周期,适配自动化提取 + 检测试剂盒 + 菌株 + qPCR 仪、外源因子一体机等多种配置,满足不同企业的个性化检测需求,获得市场认可。
病毒收获液、抗体 UPB 等中间产物需逐批检测支原体,把控生产过程质量。
支原体是一类无细胞壁的原核微生物,直径只有 0.1-0.8 µm,常规 0.22 µm 细菌过滤器无法有效去除,其污染在细胞培养领域极为普遍。这类微生物会严重干扰培养细胞的表型特征与正常生长,给生物制品质量安全带来极大隐患,且与细菌、真菌污染不同,支原体污染通常不会导致培养液浑浊或细胞可见病变,需通过专业方法检测。基于此,FDA、WHO 等全球监管机构及 USP、EP、ChP 等主流药典均明确要求,对测试细胞库(主细胞库、工作细胞库)、下游细胞培养物、终产品及对照细胞等关键环节开展支原体污染筛查,确保生物制品生产全流程安全可控。
支原体兼具胞内胞外生存特性,检测需裂解细胞而非只取上清,避免漏检胞内污染。安徽生物制品支原体检测使用性验证
支原体检测过程中,需严格遵循 “先阴后阳” 操作原则,避免交叉污染。河南复杂基质支原体检测试剂盒
长期以来,支原体检测主要依赖培养法和指示细胞法,且法规通常要求两种方法同时使用,但这两类方法存在明显短板——培养法检测周期长达 28 天,指示细胞法也需较长时间等待结果。随着细胞疗法药物快速发展,其上市周期短、货架期有限的特点,使得传统方法难以满足药物放行的时效要求。核酸扩增技术(NAT)尤其是荧光探针 qPCR 检测方法的出现,凭借检测速度快、特异性强的优势,成为支原体检测的理想替代方案。作为替代方法,NAT 检测需通过严格验证以达到法规要求的灵敏度:检测限达到 10CFU/mL 可替代培养法,达到 100CFU/mL 可替代指示细胞培养法,从而实现快速且可靠的支原体筛查。
河南复杂基质支原体检测试剂盒