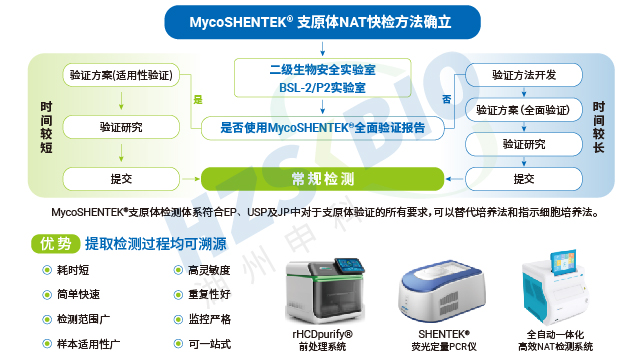
全球主流药典均认可 NAT 法作为支原体检测的替代方法,但明确要求需经过充分验证并证明与传统方法的可比性。欧洲药典(EP)2.6.7 规定 NAT 法可作为替代方法,需开展供试品抑制性验证;美国药典(USP)63&77 要求替代方法需与培养法、指示细胞培养法均具备可比性,且 USP<77> 专门针对支原体 NAT 法的验证与检测制定了规范;日本药典(JP)G3-14-170 允许经适当验证后的 NAT 法替代传统方法;中国药典 3301 及相关指导原则明确,CAR-T、干细胞等新型产品可采用 NAT 法,但需充分验证其最低检出限不低于药典方法,早期需与药典方法并行使用。此外,各国法规均强调,NAT 法的选择、开发与应用需基于科学依据,充分考虑培养基、生产工艺、潜在污染菌株等因素。
支原体污染来源多样,包括人源、动物源及环境,需多方位监测。河北疫苗产品支原体检测国产替代
培养基的科学选择与合规使用是支原体培养法检测成功的基础,湖州申科按 USP 标准明确了三类推荐培养基的适用场景。Hayflick Media 用于支原体一般性检测,Frey Media 专门针对滑液囊支原体检测,Friis Media 则适用于非禽类支原体检测。为确保少量支原体(约 100cfu 或 100ccu)不被遗漏,需使用足够数量的固体与液体培养基开展检测;若选用其他替代培养基,必须严格符合 USP 标准要求。此外,每批培养基均需进行针对性的微生物检测(即营养特性测试),通过标准化的质量把控,避免因培养基性能缺陷导致检测失效,为后续检测流程提供稳定可靠的基础条件。
江西干细胞产品支原体检测快速检测高浓度质粒样品在进行支原体检测时,需通过浓缩离心预处理,提升支原体检出率。
湖州申科围绕 USP 支原体培养法,提供技术服务,满足生物制剂企业的合规需求。服务内容包括两项:一是支原体培养法样品检测服务,严格遵循 USP 63 标准流程执行,确保检测结果合规有效;二是支原体培养法样品适用性验证服务,针对供试品特性开展定制化验证,保障检测方法与样品的适配性。所有检测与验证完成后,公司将出具标准化报告,报告可直接用于项目申报、审批、工艺优化等场景,为企业产品质量控制提供依据。通过专业的技术服务,帮助企业规避合规风险,确保生物制剂生产全流程的质量与安全性,助力企业高效推进产品研发与上市进程。
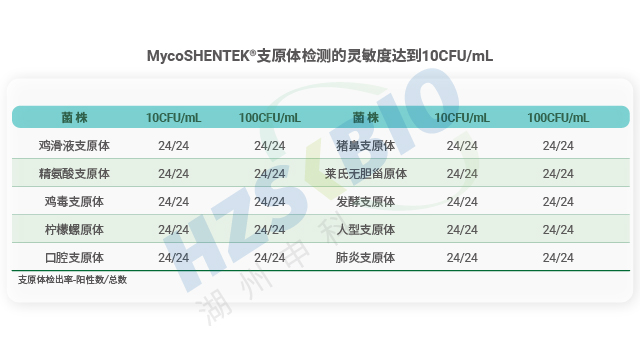
MycoSHENTEK® 支原体 qPCR 检测试剂盒(2G)完全符合 EP 2.6.7 的全部验证要求,其检测灵敏度、特异性、耐用性均按药典标准完成完整性能验证,具备替代培养法和指示细胞培养法的合规资质。该试剂盒针对新型生物制品的检测痛点优化升级,经多种支原体菌株验证,灵敏度稳定达到 10 CFU/mL,满足法规对替代培养法的要求。同时,产品遵循 ISO13485 体系认证和 GMP-like 生产标准,可提供完整的验证报告、质检报告及菌株溯源文件,全程贴合各国药典监管要求,为企业合规检测提供坚实支撑。
湖州申科支原体检测快速版试剂盒 2-2.5 小时出结果,适配紧急放行,满足细胞疗法时效需求。
菌株质量是支原体检测 NAT 方法验证合规的关键,GC/CFU 比(基因组拷贝数与菌落形成单位比值)是关键控制指标。支原体存在聚集特性,单一 CFU 可能对应多个菌体,且 DNA 复制与细胞分裂不同步,部分菌体无法形成菌落但会释放 DNA,导致 GC/CFU 比波动大(研究报道 0.1 CFU 对应 30-500 个基因组拷贝)。若使用高 GC/CFU 比菌株,会高估检测限、导致方法灵敏度不足,因此法规明确要求菌株 GC/CFU 比<10。2024 年 EDQM 36.1 草案规定参考品 GC/CFU 比应小于 10,USP 77 征求意见稿要求表征菌株 GC/CFU 比、建立菌株库 CFU 与核酸拷贝数关系,JP G3-14-170 也对菌株质量提出明确要求,凸显合规菌株选择的重要性。
USP<77> 草案新增支原体 NAT 法方法学验证章节,明确检测限需≥24 次数据支撑统计分析。河北疫苗产品支原体检测国产替代
支原体兼具胞内胞外生存特性,检测需裂解细胞而非只取上清,避免漏检胞内污染。河北疫苗产品支原体检测国产替代
细胞和基因治疗领域正加速发展,国内以 CAR-T、间充质干细胞、AAV 基因治疗等新型生物制品势头正盛。这类产品与传统制药差异明显,给支原体检测带来全新挑战:批产量小但批次多,多数待检测样品含高达 10⁷个活细胞,且基质复杂如高蛋白、全血、高浓度质粒等。更关键的是,新型生物制品终末灭菌难度极大,需从起始材料、原物料到全工艺过程严格控污,而支原体污染隐蔽性强、危害大,成为质量安全控制的主要痛点,也推动着检测方法向更高效、抗干扰的方向升级。
河北疫苗产品支原体检测国产替代