eCTD的实施为监管机构和企业带来了多重机遇。电子化申报资料能够极大地加速审评效率,减少人为判断错误和数据混淆的情况,从而提高审评的准确性和速度。同时,eCTD带来的数据标准化机遇使得全球监管机构的资料内容和电子格式得以统一,有助于在不同监管机构之间进行数据传输和共享。这对于提升全球监管效率和行业研发效率具有重要意义。 此外,eCTD的实施还促进了国际合作,构建了全球监管的底层大数据基础。对于企业而言,eCTD提供了一个规范化的研发活动模板,有助于降低与监管机构沟通的成本,提高申报效率。特别是对于国内的生物技术企业而言,eCTD的实施更是具有重要意义,有助于这些企业更好地走向国际市场。然而,中小企业在享受这些机遇的同时,也面临着技术和成本压力。eCTD的实施需要专门的团队进行系统维护和开发,这对于中小企业来说是一笔不小的开支。同时,数据安全问题也是企业关注的焦点。 此次CDE扩大eCTD实施范围对行业而言是一个积极的风向标。短期内,企业面临的挑战包括适应更高要求的技术规范并提高文件质量、和eCTD出版系统的磨合以及进行eCTD知识的跨职能培训等。 美国eCTD验证标准相关技术支持。苏州中国eCTD找哪家
eCTD生命周期管理与变更提交:欧盟要求eCTD申报资料覆盖药品全生命周期,包括提交、补充申请及实质性变更。例如,增成员国需提交“附加成员国序列”,审评时间约52-83天;重大变更(如生产工艺调整)需创建序列并通过CTIS平台更模块3和模块1的GMP证明。技术验证工具(如EDQM推荐的检查软件)需在每次提交前运行,确保XML骨架文件与PDF书签层级符合规范。此外,电子签章需符合《欧盟电子签名法》,并在模块1中明确标注法律效力。欧洲通用提交门户(Common European Submission Portal,CESP)是欧盟及成员国药品监管机构间用于电子化提交申报资料的重要平台。以下是关于CESP的详细介绍: CESP是由欧盟药品监管部门负责人网络(HMA)合作开发的在线交付系统,旨在为药品注册申请者、利益相关方和监管机构之间提供统一、安全的电子提交通道。其设计初衷是简化跨国申报流程,允许通过单一门户向多个欧洲国家的药监部门同时提交申请,避免了重复操作。吴江区CDE eCTD性价比高美国eCTD注册外包相关技术支持。
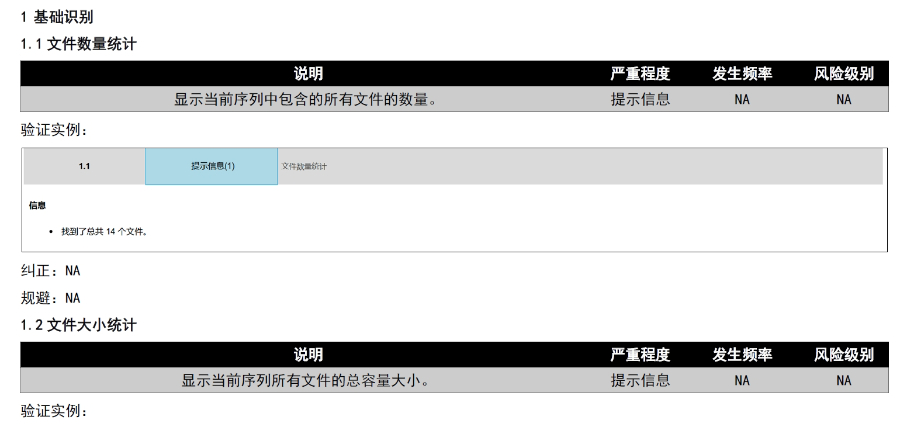
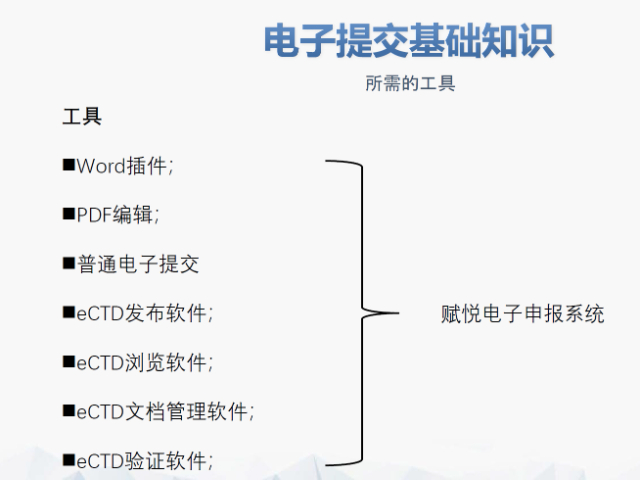
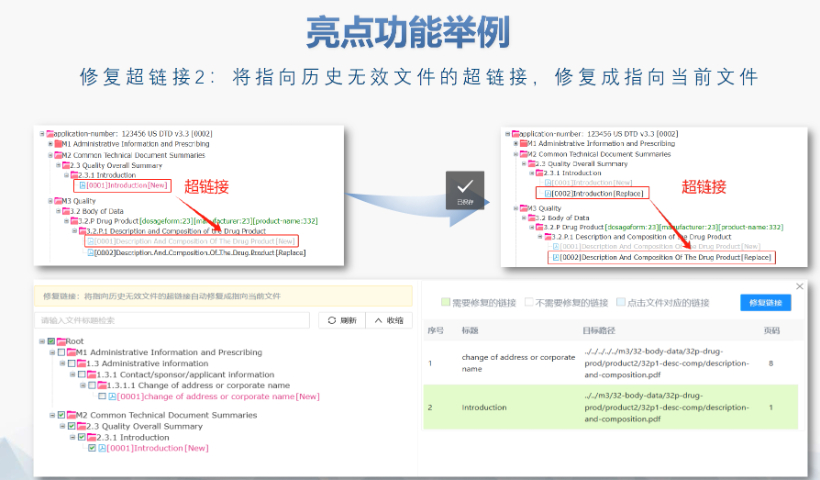
文件生命周期管理:eCTD支持文件替换(Replace)、删除(Delete)等操作,而非增文件。例如,更临床研究方案时需用Replace操作覆盖旧版本。基线提交(Baseline Submission)可用于补充历史纸质资料,但需在封面函中声明无内容变更。 临床数据与研究标签文件(STF):模块4和5中的研究数据需通过STF(Study Tagging Files)引用,确保数据与文档关联。FDA要求数据集(如SAS XPORT格式)能置于模块3-5,且单个文件超过4GB需拆分。2022年统计显示,58%的ANDA因研究数据技术拒绝标准(TRC)错误被拒。 电子签名与表格要求:FDA表格(如356h、1571)需使用数字签名,PDF文件禁止加密或设置编辑限制。电子签名需符合21 CFR Part 11规范,确保身份验证、不可否认性和数据完整性。 外包服务与系统解决方案:赋悦科技累计提交超2000份eCTD申请,外包可降低40%人工错误率。
紧急申报与特殊通道:FDA设置紧急申报通道(如Pre-EUA和EUA),允许在公共卫生事件中快速提交资料。此类申请需在模块1.19注明特殊标识,并通过ESG加急处理。 eCTD版本兼容性与过渡策略:eCTD 4.0支持向前兼容,允许v3.2.2文件无缝过渡。企业需在2024年前完成系统升级,确保XML到HL7 RPS的格式转换。过渡期间需同时维护旧版本系统。 区域差异与全球化协调:美国模块1要求严格,如UUID标识符和组合申请支持,而欧盟侧重文件引用合规性。FDA与PMDA、EMA通过ICH框架协调eCTD标准,但区域特殊性仍需针对性适配。 行业影响与长期价值:eCTD不是技术升级,更是全球药品监管一体化的驱动力。其标准化、可追溯性和效率提升,推动了跨国多中心试验的协同申报,加速创药上市进程。欧盟eCTD注册咨询相关技术支持。

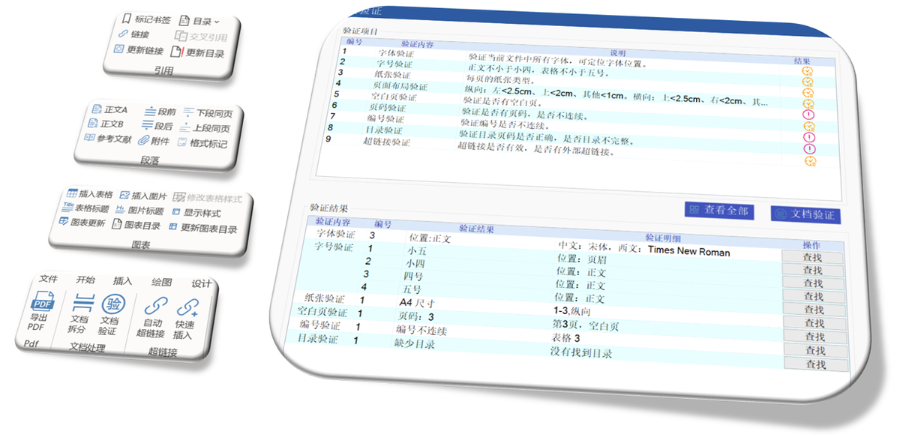
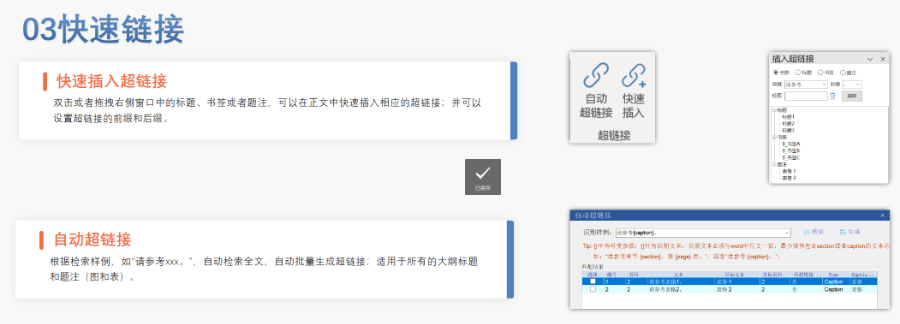
内容与格式检查Word预处理:需检查拼写、缩略语、单位格式(如),设置多级列表自动编号(如),统一字体(宋体/TimesNewRoman)和段落格式。重复内容处理:相同剂型不同规格可共用模块3,但需区分包装系统(如、)。外文资料:中文在前、原文在后,参考文献需中英文对照并建立跨网页链接。使用符合ICH标准的eCTD编辑器自动生成XML骨架和MD5校验值,拖拽PDF文件构建结构树。序列管理:序列号从0000开始递增,每次提交需更新序列,生命周期状态(New/Replace/Delete)需在XML中明确标注。验证与递交:确保无验证错误(如书签缺失、超链接断链),通过ESG等电子通道传输,光盘封面需包含申请号和序列号。全生命周期管理版本:通过软件实现网页签入/签出、审批流程,支持历史版本追溯。变更管理:增补(Append)和替换(Replace)需关联原始序列,删除(Delete)需彻底移除无效文件。 eCTD注册咨询相关技术支持。浙江eCTD文件如何制作
美国ESG电子提交通道申请相关技术支持。苏州中国eCTD找哪家
多国审评程序与eCTD递交途径的适配:欧盟药品审评程序包括集中(CP)、分散(DCP)、互认(MRP)和国家程序(NP),eCTD需适配不同程序的递交要求。例如: 集中审评程序(CP):通过EMA的eSubmission Gateway提交,审评时限约240个工作日,eCTD需包含完整的模块1-5及多语言标签文件。 分散审评程序(DCP):需通过CESP(欧盟共同提交门户)递交,参考成员国(RMS)主导审评,eCTD需支持多国同步评估的模块化拆分。 互认程序(MRP):已授权成员国作为RMS,eCTD需包含基线序列(Baseline Sequence 0000)以整合历史审评数据,并通过CMDh协调分歧。苏州中国eCTD找哪家