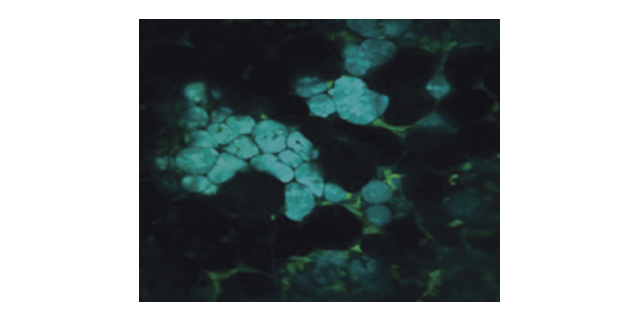
后续实验使用碘化丙啶(PI)来指示细胞在7、8、9和10分钟的延时观察后的损伤情况,来验证该光学系统对活细胞长期观察的适用性。在观察期间,88个焦点以100毫秒的曝光时间,曝光间隔1s照射样品,激发强度为3.21×104W/cm2,激发波长为525nm,使用前文提到的60×物镜及1.0AU孔径,图5(a)-(d)为引入PI的成像图,(e)-(h)为相应的相应衬度图。改变激发条件为每照射500ms间隔5s,得到相应的(i)-(p)。由图像可知,延时观察小于8分钟的情况下不造成可见细胞损伤,对于实际3D延时成像,由于焦平面是移动的,所以预期细胞存活时间会更长,可见这是一种在3D在体延时成像中具有很大优势的成像方案。双光子显微镜使用高能量锁模脉冲激光器。2PPLUS双光子显微镜成像视野是多少
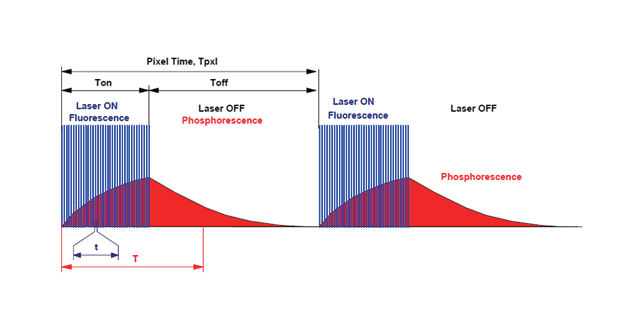
在深度组织中以较长时间对活细胞成像,双光子显微镜是当前之选。双光子和共聚焦显微镜都是通过激光激发样品中的荧光标记,使用探测器测量被激发的荧光。但是,共聚焦一般使用单模光纤耦合激光器,通过单光子激发荧光,而双光子使用飞秒激光器,通过几乎同时吸收两个长波光子激发荧光。下面是两种技术的对比图。双光子激发荧光的主要优势:双光子比共聚焦使用的更长的波长,所以对组织的损伤更小且穿透更深。共聚焦的成像深度一般为100微米,双光子则能达到250到500微米,甚至超过1毫米。另外,同时吸收两个光子意味只有较强度聚焦点处能被激发,所以不会损伤焦平面之外的组织,并且生成更清晰的图像。美国激光荧光双光子显微镜成像原理双光子显微镜放大倍数是多少?

其实电子显微镜相比光学显微镜的重要优势或意义不在于放大倍数,而在于超高的分辨率。这两者是不同的。一般来说,观察时,除了放大物体外,还需要将其与其他相邻物体区分开来。如果两个相邻粒子的图像在光学显微镜下,即使放大很大程度,也可能看到两个相交的亮点(艾里斑),没有明显的边界(更不用说细节了),说明分辨率不够。没有分辨率谈放大是没有意义的。光学显微镜的分辨率极限是阿贝极限,大约是光波波长的一半。通常称之为光学显微镜的放大极限,但准确的说应该叫分辨率极限。原因是光的衍射,根本原因是光的波粒二象性。电子衍射实验证明了电子的波动性,所以在电子显微镜中用电子代替光是可能的。电子显微镜也有很多种,被摄体像REM。也有根据衍射规律观察的电子显微镜,如低能电子衍射(LEED)和透射电子显微镜(TEM)。两者主要用于观察晶体,根据晶体的周期特性在倒易空间产生衍射像,借助埃尔沃德球或傅里叶变换将其变换到实空间,即可得到真实的晶体表面像。
而配合了双光子激发技术,激光共聚扫描显微镜则能更好得发挥功效。那么,什么是双光子激发技术呢?在高光子密度的情况下,荧光分子可以同时吸收2个长波长的光子使电子跃迁到较高能级,经过一个很短的时间后,电子再跃迁回低能级同时放出一个波长为长波长一半的光子(P=h/λ)。利用这个原理,便诞生了双光子激发技术。双光子显微镜使用长波长脉冲激光,通过物镜汇聚,由于双光子激发需要很高的光子密度,而物镜焦点处的光子密度是比较高的,所以只有在焦点处才能发生双光子激发,产生荧光,该点产生的荧光再穿过物镜,从而被光探头接收,从而达到逐点扫描的效果。双光子显微镜在各领域研究中已有许多成功实例。

许多生物医学成像方式,无论是单光子(共聚焦)或多光子(双光子),都使用激光作为光源,并需要兼容的荧光染料。荧光染料有自己的激发波长,它们可以被单个光子以该激发波长的光子能量激发(E=hv=h*c/λ);或者是两个几乎同时到达的光子,但每个光子的能量约为单光子能量的一半,即双波长(0.5E->2λ)。前者是单光子显微镜原理,后者是双光子显微镜原理。在对同一种荧光染料进行成像时,双光子与单光子相比可以使用约两倍波长,因此双光子的散射较小(波长较长,散射较小),可以更深入地渗透到组织中。双光子显微镜型号有哪些?国外2PPLUS双光子显微镜授权公司
双光子显微镜可以用于局部微蚀镭射磨皮后的胶原重塑的检测。2PPLUS双光子显微镜成像视野是多少
实验从理论和实验上评估了多焦点v2PE显微镜的空间分辨率,并与单光子荧光显微镜进行了对比,实验中v2PE的激发波长为521nm,使用放大倍率为100倍的物镜,尺寸为0.6AU,对直径100nm的荧光颗粒进行了测试性成像,共获得40幅不同采样深度的图像合成为三维图像。图像在横向和纵向的半高全宽分别是177nm和297nm,这些值接近显微镜的理论分辨率。后续还利用软件模拟从理论上研究了多焦点v2PE显微技术的空间分辨率,模拟计算显示v2PE点扩散函数(PSF)的横向半高宽与单光子激发荧光(1PE)相似,轴向的半高宽较1PE减少,可以提高空间分辨率。2PPLUS双光子显微镜成像视野是多少