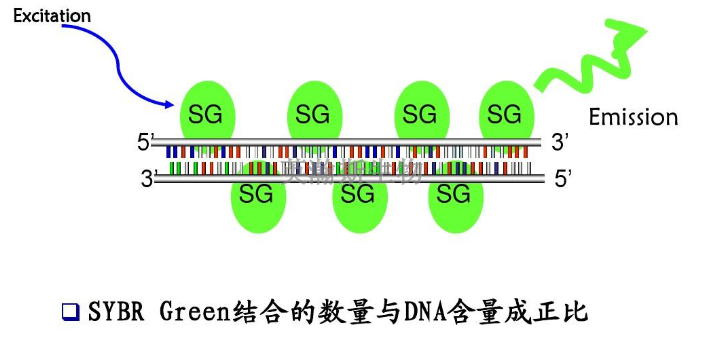
原位PCR就是在组织细胞里进行PCR反应,它结合了具有细胞定位能力的原位杂交和高度特异敏感的PCR技术的优点,是细胞学科研与临床诊断领域里的一项有较大潜力的新技术。原位PCR是Hasse等于1990年建立的,实验用的标本是新鲜组织、石蜡包埋组织、脱落细胞、血细胞等。其基本方法为:1、固定组织或细胞:将组织细胞固定于预先用四氟乙烯包被的玻片上,并用多聚甲醛处理,再灭活除去细胞内源性过氧化物酶。2、蛋白酶K消化处理:用60ug/ml的蛋白酶K将固定好的组织细胞片55℃消化处理2h后,96℃2min以灭活蛋白酶K。3、PCR扩增:在组织细胞片上,加PCR反应液,覆盖并加液体石蜡后,直接放在扩增仪的金属板上,进行PCR循环扩增。有的基因扩增仪带有专门用于原位PCR的装置。4、杂交:PCR扩增结束后,用标记的寡核苷酸探针进行原位杂交。5、显微镜观察结果。荧光定量PCR检测原理是什么?甘肃荧光定量pcr公司
PCR的特异性影响因素有很多,在此我们作一归纳,供大家参考:①退火步骤的严格性:提高退火温度可以减少不匹配的杂交,从而提高特异性。②减短退火时间及延伸时间可以减少错误引发及错误延伸。③引物二聚体是较常见的副产品,降低引物及酶的浓度也可以减少错误引发,尤其是引物的二聚化。④改变mgcl2(有时kcl)浓度可以改进特异性,这可能是提高反应严格性或者对taq酶的直接作用。一般认为PCR产物应在48h以内完成电泳检测,有些比较好于当日电泳检测,大于48h后带型就会出现不规则,甚至消失。湖北靠谱pcr实验室pcr实验失败的原因有哪些?
PCR反应五要素:参加PCR反应的物质主要有五种即:引物(PCR引物为DNA的片段,细胞内DNA复制的引物为一段RNA链)、酶、dNTP、模板和缓冲液(其中需要Mg2+)。引物有多种设计方法,由PCR在实验中的目的决定,但基本原则相同。PCR所用的酶主要有两种来源:Taq和Pfu,分别来自两种不同的噬热菌。其中Taq扩增效率高但易发生错配。Pfu扩增效率弱但有纠错功能。所以实际使用时根据需要必须做不同的选择。模板即扩增用的DNA,可以是任何来源,但有两个原则,1号纯度必须较高,第二浓度不能太高以免抑制。缓冲液的成分较为复杂,除水外一般包括四个有效成分:缓冲体系,一般使用HEPES或MOPS缓冲体系;一价阳离子,一般采用钾离子,但在特殊情况下也可使用铵根离子;二价阳离子,即镁离子,根据反应体系确定,除特殊情况外不需调整;辅助成分,常见的有DMSO、甘油等,主要用来保持酶的活性和帮助DNA解除缠绕结构。
PCR实验技术中cDNA第二链的合成方法有以下几种:1、自身引导法合成的单链cDNA3'端能够形成一短的发夹结构,这就为第二链的合成提供了现成的引物,当***链合成反应产物的DNA:RNA杂交链变性后利用大肠杆菌DNA聚合酶ⅠKlenow片段或反转录酶合成cDNA第二链,***用对单链特异性的S1核酸酶消化该环,即可进一步克隆。但自身引导合成法较难控制反应,而且用S1核酸酶切割发夹结构时无一例外地将导致对应于mRNA5'端序列出现缺失和重排,因而该方法目前很少使用。2、置换合成法该方法利用链在反转录酶作用下产生的cDNA:mRNA杂交链不用碱变性,而是在dNTP存在下,利用RNA酶H在杂交链的mRNA链上造成切口和缺口。从而产生一系列RNA引物,使之成为合成第二链的引物,在大肠杆菌DNA聚合酶Ⅰ的作用下合成第二链。该反应有3个主要优点:(1)非常有效;(2)直接利用链反应产物,无须进一步处理和纯化;(3)不必使用S1核酸酶来切割双链cDNA中的单链发夹环。pcr内标不出来的原因是什么?
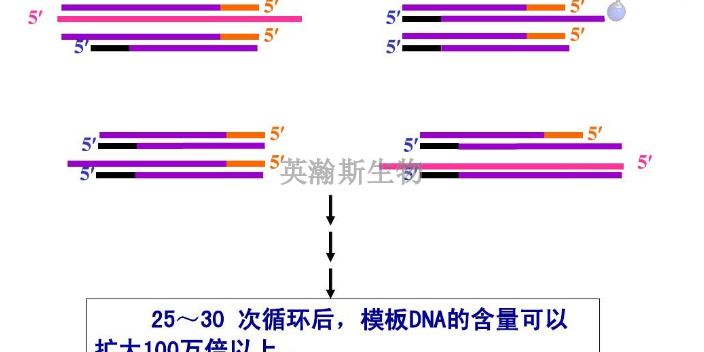
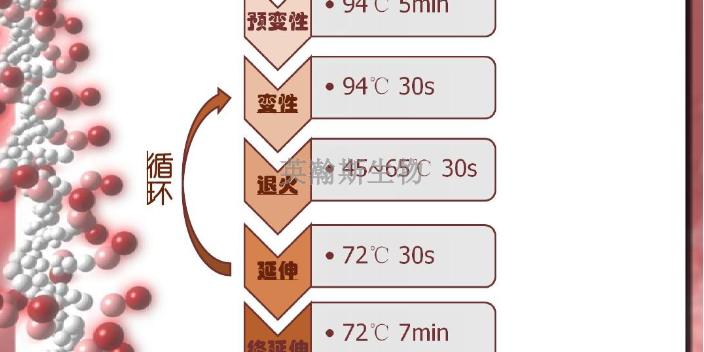
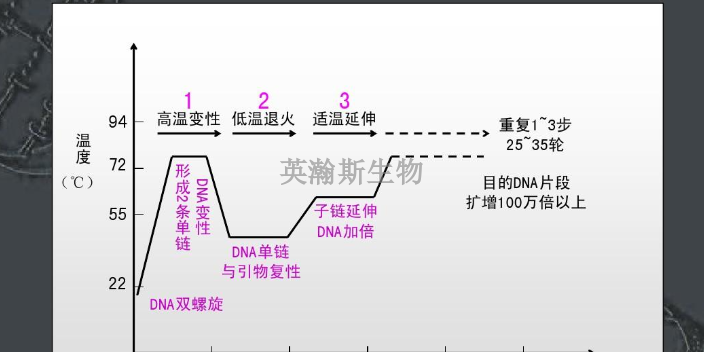
PCR技术的基本原理类似于DNA的天然复制过程,DNA的半保留复制是生物进化和传代的重要途径。其特异性依赖于与靶序列两端互补的寡核苷酸引物。PCR由变性-退火-延伸三个基本反应步骤构成:①模板DNA的变性:模板DNA经加热至93℃左右一定时间后,使模板DNA双链或经PCR扩增形成的双链DNA解离,使之成为单链,以便它与引物结合,为下轮反应作准备;②模板DNA与引物的退火(复性):模板DNA经加热变性成单链后,温度降至55℃左右,引物与模板DNA单链的互补序列配对结合;③引物的延伸:DNA模板-引物结合物在72℃、DNA聚合酶(如TaqDNA聚合酶)的作用下,以dNTP为反应原料,靶序列为模板,按碱基互补配对与半保留复制原理,合成一条新的与模板DNA链互补的半保留复制链,重复循环变性-退火-延伸三过程就可获得更多的“半保留复制链”,而且这种新链又可成为下次循环的模板。每完成一个循环需2~4分钟,2~3小时就能将待扩目的基因扩增放大几百万倍英瀚斯生物,可承接各类荧光定量pcr检测。江苏荧光定量pcr怎么样
英瀚斯生物,确保pcr检测结果真实性。甘肃荧光定量pcr公司
PCR的假阳性主要原因之一出现非特异性扩增带:PCR扩增后出现的条带与预计的大小不一致,或大或小,或者同时出现特异性扩增带与非特异性扩增带。非特异性条带的出现,其原因:一是引物与靶序列不完全互补、或引物聚合形成二聚体。二是Mg2+离子浓度过高、退火温度过低,及PCR循环次数过多有关。其次是酶的质和量,往往一些来源的酶易出现非特异条带而另一来源的酶则不出现,酶量过多有时也会出现非特异性扩增。其对策有:必要时重新设计引物。减低酶量或调换另一来源的酶。降低引物量,适当增加模板量,减少循环次数。适当提高退火温度或采用二温度点法(93℃变性,65℃左右退火与延伸)。PCR的假阳性主要原因之一出现片状拖带或涂抹带:PCR扩增有时出现涂抹带或片状带或地毯样带。其原因往往由于酶量过多或酶的质量差,dNTP浓度过高,Mg2+浓度过高,退火温度过低,循环次数过多引起。其对策有:减少酶量,或调换另一来源的酶。②减少dNTP的浓度。适当降低Mg2+浓度。增加模板量,减少循环次数。甘肃荧光定量pcr公司
南京英瀚斯生物科技有限公司主要经营范围是医药健康,拥有一支专业技术团队和良好的市场口碑。公司业务分为实验外包,动物模型构建,细胞分子实验,病理检测等,目前不断进行创新和服务改进,为客户提供良好的产品和服务。公司注重以质量为中心,以服务为理念,秉持诚信为本的理念,打造医药健康良好品牌。在社会各界的鼎力支持下,持续创新,不断铸造***服务体验,为客户成功提供坚实有力的支持。